 |
 |
 |
 |
 |
|---|
![]() Le séquençage des génomes
Le séquençage des génomes
Depuis que l'ADN a été reconnu comme support de l'information génétique, bien des biologistes ont rêvé de séquencer l'intégralité du génome d'un être vivant. En effet, connaître l'enchaînement complet des bases nucléotidiques qui constituent un génome, c'est connaître toute l'information nécessaire à la vie (du moins en théorie). Un tel rêve est longtemps resté inaccessible, pour des raisons technologiques et financières. Ce n'est que récemment, avec la mise en place des Programmes Génome, que des génomes entiers ont pu être entièrement déchiffrés.
- 1. Les choix stratégiques
- 1.1 Approches utilisées pour le séquençage à grande échelle
- 1.2 Organismes séquencés
- 1.3 Identification des gènes
- 2. Génomes procaryotes
- 2.1 Structure chromosomique
- 2.2 Organisation des gènes
- 2.3 Séquences non codantes
- 2.4 Retombées médicales et commerciales
- 3. Génomes des modèles eucaryotes
- 3.1 Structure des chromosomes
- 3.2 Identification des gènes
- 3.3 Fonctions des gènes reconnus ou prédits
- 3.4 Régions non codantes
- 4. Génome humain
- 4.1 Chromosomes humains
- 4.2 Identification des gènes
- 4.3 Séquences répétées
- Conclusion
1. Les choix stratégiques
1.1 Approches utilisées pour le séquençage à grande échelle
Deux approches différentes sont utilisées pour le séquençage massif. La première consiste à diviser le travail entre une multitude de laboratoires, auxquels sont alloués les crédits nécessaires au séquençage de la région qui leur est attribuée. C'est par exemple ainsi que furent entièrement séquencés les génomes des Procaryotes B. subtilis (46 laboratoires; 1997) ou Xylella fastidiosa (34 laboratoires; 2000). Le premier chromosome d'un organisme eucaryote à être entièrement séquencé a aussi suivi cette voie: la séquence du chromosome III de la levure (315 kb) a été obtenue en 1991 par un consortium de 35 laboratoires européens. Ce succès a par la suite mené au séquençage de plusieurs autres chromosomes de la levure.
La seconde approche consiste à concentrer le travail dans des Genome Centers où toutes les étapes sont réalisées sur une grande échelle de production. Cette concentration permet une rentabilité optimale des différentes étapes, ainsi que le développement de nouvelles technologies. La plupart de ces centres se consacrent au séquençage d'un génome particulier. C'est l'option qui domine aujourd'hui, et qui a produit d'importants ensembles de séquences.
Les techniques utilisées pour le séquençage intensif ont bien sûr évolué suivant une tendance à l'automatisation de plus en plus poussée. Pour le premier chromosome de la levure, moins de 10% des séquences avait été obtenu sur séquenceurs automatiques. Mais la majorité des autres travaux ont mis en évidence l'importance croissante des étapes d'automatisation, et l'utilisation quasi exclusive de séquenceurs automatiques. Ces machines minimisent les erreurs susceptibles d'être introduites par une intervention humaine, et augmentent donc la précision du séquençage.
1.2 Organismes séquencés
Des bactéries telles que E. coli, B. subtilis, Agrobacterium tumefaciens ou Lactococcus lactis ont été séquencées en raison de leur importance dans le domaine de la recherche fondamentale, ou en raison de leur utilisation industrielle, en particulier dans le domaine agro-alimentaires.
Un nombre important de génomes de Procaryotes pathogènes ont été séquencés (tableau 1). La priorité a essentiellement concerné des pathogènes de l'homme, puisque la moitié de nos maladies est d'origine bactérienne. Ont donc été choisis H. influenzae (responsable de bronchites, otites, fig 1), Chlamydia trachomatis (infections génitales, pulmonaires, oculaires), Chlamydia pneumoniae (pharyngites, bronchites, pneumonies), V. cholerae (choléra), Mycoplasma genitalium (tractus génital), Mycoplasma pneumoniae (pneumonies), Helicobacter pylori (ulcères), Mycobacterium tuberculosis (tuberculose), Mycobacterium leprae (lèpre), Treponema pallidum (syphilis), Rickettsia prowazekii (typhus), B. burgdorferi (maladie de Lyme), Yersinia pestis (peste), Neisseria gonorrhoeae (gonorrhée), Neisseria meningitidis (méningite), Listeria monocytogenes (méningites, avortements), Campylobacter jejuni (syndrome de Guillain-Barré), E. coli O157:H7 (E. coli virulente), Ureaplasma urealyticum (pathogène urogénital), Shigella flexneri (dysenterie et réaction inflammatoire), Pseudomonas aeruginosa (infectant en particulier les patients atteints de mucoviscidose, ou les individus immunodéficients)... Certains pathogènes d'animaux ou végétaux ont aussi représentés de tels projets, comme par exemple X. fastidiosa, phytopathogène affectant en particulier les orangers, Xanthomonas campestris, pathogène de divers crucifères, ou Ralstonia solanacearum, capable de contaminer plus de 200 espèces végétales.
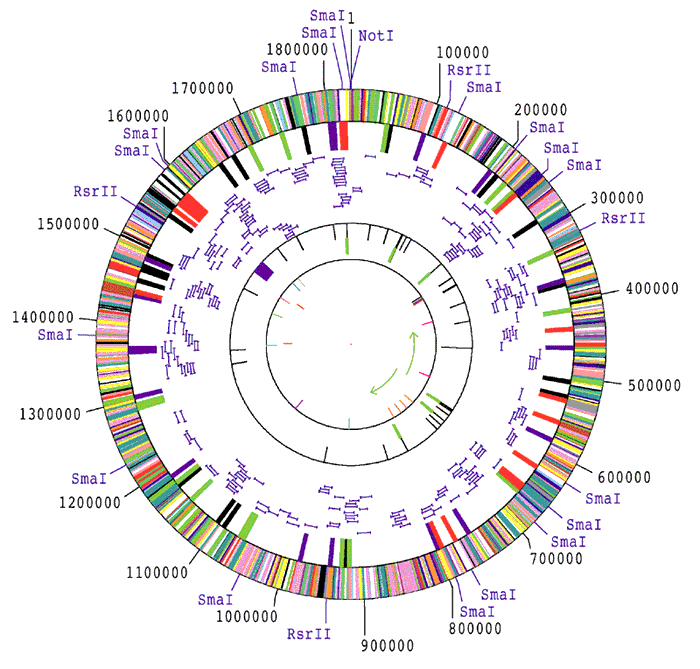
Figure 1. Génome de Haemophilus
influenzae.
Premier cercle: sites de restriction (enzymes Not I, Sma I, Rsr II) et numérotation des bases.
Deuxième cercle: ensemble des ORF, colorées selon leur fonction.
Troisième cercle: régions riches en [G+C], ou riches en [A+T].
Des génomes d'Archébactéries ont aussi été séquencés. Plusieurs de ces espèces vivent à haute température, ou présentent des métabolismes particuliers (production de méthane, réduction de sulfates...) Ces analyses permettraient donc de révéler des enzymes industriellement intéressants.
Enfin, certaines bactéries ont été séquencées en raison de particularités biologiques: c'est le cas de Buchnera, vivant en symbiose avec des Insectes, ou de D. radiodurans, capable de survivre dans des milieux fortement irradiés.
Tableau 1. Génomes procaryotes totalement séquencés. Le premier groupe comprend des exemples de génomes d'Archébactéries, le second ceux d'Eubactéries.
| Espèce | taille du génome (pb) | nombre d'ORF | ORF nouvelles (et pourcentage) | taux de [G+C] | fraction codante | nombre d'ARNt |
|---|---|---|---|---|---|---|
| Aeropyrum pernix | 1.669.695 | 2.694 | 1.536 (57%) | 56% | 89% | 47 |
| Archaeoglobus fulgidus | 2.178.400 | 2.436 | 1.290 (53%) | 48,5% | 92% | 46 |
| Halobacterium NRC-1 | 2.571.010 | 2.682 | 1.615 (60%) | 68% | nd | 47 |
| Methanococcus jannaschii | 1.664.970 | 1.738 | 1.078 (62%) | 31% | 88% | 37 |
| Methanosarcina acetivorans | 5.751.492 | 4.524 | 2298 (51%) | 42% | 74% | nd |
| Pyrococcus abyssi | 1.765.118 | 1.765 | 865 (49%) | 45% | 91% | 46 |
| Pyrococcus horikoshii | 1.738.505 | 2.061 | 859 (42%) | 42% | 91% | 46 |
| Thermoplasma acidophilum | 1.564.905 | 1.509 | 686 (45%) | 46% | 87% | 45 |
| Aquifex aeolicus | 1 551 335 | 1.512 | 663 (44%) | 43% | 94% | 44 |
| Bacillus subtilis | 4.214.810 | 4.100 | 1.721 (42%) | 43,5% | 97% | 88 |
| Borrelia burgdorferi | 910.725 | 853 | 353 (41%) | 29% | 93% | 34 |
| Buchnera | 640.681 | 583 | 111 (19%) | 26% | 88% | 32 |
| Campylobacter jejuni | 1.641.481 | 1.654 | 367 (22%) | 31% | 94% | nd |
| Chlamydia trachomatis serovar D | 1.042.519 | 894 | 290 (32%) | 41% | nd | 37 |
| Chlorobium tepidum | 2.154.946 | 2.288 | 1.071 (47%) | 56,5% | 89% | 50 |
| Deinococcus radiodurans | 3.284.156 | 3.187 | 1.694 (53%) | 67% | 91% | 49 |
| Escherichia coli | 4.639.221 | 4 288 | 1.632 (38%) | 51% | 88% | 86 |
| Fusobacterium nucleatum | 2.174.500 | 2.067 | 673 (33%) | 27% | 90% | 47 |
| Haemophilus influenzae | 1.830.127 | 1.743 | 732 (42%) | 38% | 87% | 54 |
| Helicobacter pylori 26695 | 1.667.867 | 1.552 | 657 (42%) | 39% | 91% | 36 |
| H. pylori J99 | 1.643.831 | 1.495 | 621 (42%) | 39% | 91% | 36 |
| Lactococcus lactis | 2.365.589 | 2.310 | 828 (36%) | 35% | 87% | 62 |
| Mycobacterium leprae | 3.268.203 | 1.604 | nd | 58% | 49,5% | nd |
| Mycobacterium tuberculosis | 4.411.529 | 3.924 | 628 (16%) | 66% | 91% | 45 |
| Mycoplasma genitalium | 580.070 | 470 | 187 (39%) | 32% | 88% | 33 |
| Mycoplasma pneumoniae | 816.394 | 677 | 344 (46%) | 40% | 89% | 33 |
| Mycoplasma pulmonis | 963.879 | 782 | 296 (38%) | 27% | 91% | 29 |
| Neisseria meningitidis (MC58) | 2.272.351 | 2.158 | 877 (41%) | 51,5% | 83% | 59 |
| Pseudomonas aeruginosa | 6.264.403 | 5.570 | 2.549 (46%) | 67% | 89% | 63 |
| Ralstonia solanacearum | 5.810.922 | 5.129 | 2.868 (56%) | 67% | 93% | 58 |
| Rickettsia prowazekii | 1.111.523 | 834 | 208 (25%) | 29% | 76% | 33 |
| Shigella flexneri | 4.607.203 | 4.434 | nd | 51% | 80% | 97 |
| Streptomyces coelicolor | 8.667.507 | 7.825 | nd | 72% | 89% | 63 |
| Streptococcus pneumoniae | 2.160.837 | 2.236 | 796 (36%) | 40% | nd | 58 |
| Synechocystis sp. | 3.573.470 | 3.168 | 1.766 (50%) | 48% | 87% | 41 |
| Thermoanaerobacter tengcongensis | 2.689.445 | 2.588 | 1.094 (42%) | 38% | 87% | 55 |
| Thermotoga maritima | 1.860.725 | 1.877 | 863 (46%) | 46% | 95% | 46 |
| Treponema pallidum | 1.138.006 | 1.041 | 464 (47%) | 53% | 93% | 44 |
| Ureaplasma urealyticum | 751.719 | 613 | 288 (47%) | 25,5% | 93% | nd |
| Vibrio cholerae | 4.033.460 | 3.885 | 1.806 (46%) | 47,5% | 88% | 98 |
| Wigglesworthia glossinidia | 697.742 | 621 | 99 (16%) | 22% | 89% | 34 |
| Xanthomonas campestris | 5.076.187 | 4.182 | 1474 (32%) | 65% | 84% | 53 |
| Xylella fastidiosa | 2.679.305 | 2.782 | 1.393 (50%) | 53% | 88% | 49 |
| Yersinia pestis | 4.653.728 | 4012 | nd | 48% | 84% | 70 |
Chez les Eucaryotes, le séquençage des génomes de la levure, de S. pombe, d'A. thaliana, du nématode, et de la drosophile sont aujourd'hui terminés (encore qu'un nombre significatif de lacunes subsistent dans le dernier cas). Ces organismes avaient été choisis en raison de la taille de leur génome, significativement plus petite que celle de l'homme, ainsi que pour leurs utilisations dans les domaines de la recherche ou économique.
1.3 Identification des gènes
Le problème crucial de l'analyse de séquences génomiques est l'identification des séquences codantes. L'identification des unités transcriptionnelles est grandement facilitée chez les Procaryotes par la possibilité d'identifier relativement facilement les promoteurs des gènes, par la quasi absence d'intron, par la possibilité de reconnaître aisément les phases ouvertes de lecture et leur terminaison, et par la faible taille des séquences intergéniques.
Deux caractéristiques compliquent radicalement l'identification des séquences codantes chez les Eucaryotes: le découpage des gènes en introns et exons, et la présence de régions intergéniques, parfois très vastes.
Chez la levure, ces problèmes restent mineurs, car seulement 5% des gènes sont morcelés en plusieurs exons, et les régions non codantes sont peu abondantes. Chez C. elegans, D. melanogaster ou A. thaliana, l'analyse des séquences est déjà beaucoup moins aisée, car les régions codantes sont en majorité fragmentées, et la fraction d'ADN non codant est importante. Cette fragmentation exon-intron est aussi la règle dans les génomes de Vertébrés, chez lesquels les régions intergéniques sont souvent très étendues.
Une identification indiscutable des unités transcriptionnelles peut être obtenue par comparaison de la séquence génomique à l'ensemble des données acquises par les programmes de séquençage d'ADNc (EST ou séquences complètes de messager, cf. chapitre 5). L'obtention d'un alignement significatif d'une séquence génomique avec une séquence ADNc permet de conclure que cette séquence est effectivement transcrite. Une autre moyen de déterminer si une séquence est codante est l'utilisation d'outils informatiques de prédiction, capables d'identifier un gène selon plusieurs critères: la présence d'une phase ouverte de lecture, de signaux d'épissage, la composition en bases. Des logiciels tels que GENSCAN (pour les séquences de mammifères), GENEFINDER (nématode), ou GENIE (drosophile), intègrent ces paramètres en un réseau du type neuronal ou utilisent des chaînes de Markov, qui optimisent la probabilité de reconnaître un gène. Mais ce type de logiciel n'est pas infaillible: environ 90% des gènes sont reconnus, mais 10% sont ignorés, et 15% des prédictions sont des faux-positifs.
L'identification de gènes peut aussi utiliser des données provenant d'un autre organisme, tels que par exemple les alignements obtenus entre la séquence génomique de C. elegans et les séquences d'EST obtenues à partir de Caenorhabditis briggsae, ou les alignements obtenus entre les séquences génomiques de Tetraodon nigroviridis ou Mus musculus dans le cas de Homo sapiens.
2. Génomes procaryotes
2.1 Structure chromosomique
Chez les Procaryotes, l'abondance en guanine et cytosine par rapport à la totalité du génome - appelé contenu en [G+C] - présente d'importantes variations selon l'espèce: entre 22% chez Wigglesworthia glossinidia et 67% chez D. radiodurans (tableau 1). Les faibles taux de [G+C] sont souvent liés à un mode de vie parasitique ou symbiotique.
La réplication du chromosome s'effectue dans les deux directions opposées divergeant à partir de l'origine de réplication. Chacune de ces "moitiés" est appelée réplichore. Chez plusieurs Eubactéries ou Archébactéries, on observe un biais dans la fréquence de nucléotides entre chaque réplichore. Chez B. subtilis, le rapport [G-C]/[G+C] s'inverse de part et d'autre de l'origine de réplication. Mais de tels biais ne sont pas toujours observés, et chez certaines espèces, la localisation de l'origine de réplication ne peut être réalisée qu'expérimentalement.
Le séquençage de génome a parfois comporté celui de plasmides: chez X. fastidiosa par exemple, ont été séquencés deux plasmides de 1.285 pb et 51.158 pb, portant respectivement 2 et 64 ORF. Des plasmides linéaires sont aussi parfois présents: B. burgdorferi contient par exemple - en plus du chromosome linéaire - sept plasmides circulaires (de 9 à 32 kb), et dix linéaires (de 17 à 56 kb). Le séquençage de mégaplasmides a d'autre part été réalisé chez divers espèces, telle que par exemple D. radiodurans (tableau 2).
Des génomes de petite taille sont fréquemment observés chez les bactéries symbiotiques, telles que Buchnera ou Wigglesworthia glossinidia, le mode de vie de ces organisme étant corrélé à leur mode de vie intracellulaire.
Tableau 2. Organisation du génome de D. radiodurans
| Taille (en nucléotides) | nombre total de gènes | Gènes de fonction inconnue | |
|---|---|---|---|
| Chromosome I | 2.648.638 | 2.633 | 1.422 |
| Chromosome II | 412.348 | 369 | 183 |
| Mégaplasmide | 177.466 | 145 | 65 |
| Plasmide | 45.704 | 40 | 24 |
| Total | 3.284.156 | 3.187 | 1.694 |
2.2 Organisation des gènes
Globalement, la fraction codante des génomes procaryotes est élevée, de l'ordre de 90% (tableau 1), variant entre 97% (B. subtilis) et 49,5% (M. leprae). La taille moyenne des gènes observée est de l'ordre de 925 pb, la plus faible étant celle de X. fastidiosa (799 pb), la plus importante celle de M. pulmonis (1.115 pb). Le nombre de gènes le plus élevé est observé chez S. coelicolor (7.825), l'autre extrême est présentée par M. genitalium, qui ne contient que 470 gènes.
Chez les Procaryotes, les unités transcriptionnelles sont fréquemment organisées en opérons. Un opéron comprend plusieurs gènes, souvent impliqués dans une même fonction physiologique. Chaque opéron est transcrit à partir d'un même promoteur, les gènes qu'il contient sont donc soumis à des phénomènes de régulation identiques. Un seul ARN messager est produit, et cet ARNm est par la suite clivé en régions correspondant à chacun des gènes, avant traduction. Le nombre de gènes par opéron est très variable: chez E. coli, environ un quart des gènes sont agencés en opérons, à l'inverse, le nombre d'opéron est très réduit chez C. jejuni.
Les gènes codant les ARNr sont par exemple souvent agencés en groupe 16S-23S-5S, constituant un opéron rrn (les régions situées entre les gènes 16S, 23S ou 5S codent souvent des ARNt). Les rrn peuvent être répétés au sein des génomes bactériens: il en existe par exemple 10 exemplaires chez B. subtilis. Mais chez d'autres espèces, telles que A. fulgidus ou M. genitalium, un seul est présent. Chez certaines espèces, les loci rrn sont organisés différemment (23S-16S-5S chez Vibrio harveyi), et il existe aussi des cas où les gènes 16S, 23S et 5S ne sont pas assemblées en opéron (A. fulgidus ou H. influenzae).
Généralement, le nombre de pseudogènes (gènes mutés, non transcrits ou non traduits) est faible, de l'ordre de 1 à 2%: chez C. jejuni sont par exemple présents 20 pseudogènes. Mais à l'inverse, M. leprae présente une fraction importante d'ADN non codant (24%) et de pseudogènes (27%).
2.3 Séquences non codantes
Les domaines non codants des génomes bactériens sont représentés par les régions intergéniques, contenant les séquences régulatrices et d'éventuelles séquences répétées, et quelques rares introns. Globalement, les séquences répétées sont beaucoup plus rares chez les Procaryotes que chez les Eucaryotes.
La distribution et l'abondance de ces éléments varient beaucoup entre espèces, souvent aussi entre diverses variétés, et aucune généralité concernant l'ensemble des Procaryotes n'a été mise en évidence. Même un génome tel que celui de M. pneumoniae, de taille pourtant très réduite (0,8 Mb), comprend 6% de séquences répétées. A l'inverse, les génomes de Buchnera ou de C. jejuni n'en présentent pratiquement aucune.
Chez E. coli, les séquences intergéniques ont une taille moyenne de 118 paires de bases. La taille des plus grandes régions atteint quelques 600 pb, et il en existe une quarantaine. Pour une vingtaine d'entre elles, il a été possible d'y reconnaître des sites de fixation de protéines régulatrices de l'expression génique. Les autres contiennent soit des séquences répétées, soit des séquences uniques dont le rôle (éventuel) reste inconnu.
Les séquences répétées en tandem comprennent un motif de 1 à 6 nucléotides, répété de 2 à quelques dizaines de fois. Les séquences répétées dispersées ne contiennent généralement pas de gènes. Dans un certain nombre de cas, leur fonction a été identifiée.
Une autre particularité connue chez les Procaryotes est leur capacité de transformation, c'est à dire d'acquisition d'un fragment d'ADN, et d'intégration dans son chromosome en lieu et place de la région homologue. Chez H. influenzae, la transformation est facilitée par l'existence d'un grand nombre de séquences spécifiques de cette espèce: ces séquences sont appelées USS (uptake signal sequences) elles sont représentées par des séquences conservées de 29 pb, comprenant un motif constant de 9 pb (5 - AAGTGCGGT - 3). Le génome de H. influenzae comporte 1.465 USS: ce nombre élevé suggère que la transformation pourrait jouer un rôle important chez cette bactérie. À l'inverse, le génome de C. trachomatis ne contient pas de séquence impliquée dans la transformation ou l'acquisition d'ADN exogène, ce qui est probablement corrélé au fait que ce parasite, isolé au sein de la cellule hôte, n'est pas amené à acquérir d'ADN externe.
La transformation a joué un rôle historique dans le domaine de la génétique, puisque c'est à partir des expériences de Griffith (1928) sur Streptococcus pneumoniae et la caractérisation du facteur transformant par Avery (1944) que l'ADN a été caractérisé comme support de l'hérédité.
2.4 Retombées médicales et commerciales
De nombreuses retombées médicales sont espérées, d'autant que les impacts des pathogènes sont très loin d'être négligeables: la syphilis touche aujourd'hui 50 millions de personnes, la lèpre 15 millions, une nouvelle pandémie de choléra est récemment apparue, chaque minute, la tuberculose atteint 10 personnes... La comparaison des génomes de différentes variétés - comme par exemple C. trachomatis MoPn et serovar D - pourrait permettre l'identification des gènes impliquées dans la spécificité pathologique. Le séquençage comparé d'espèces proches mais causant des maladies très différentes, telles que M. leprae et M. tuberculosis, ou N. meningitidis et N. gonorrhoeae, devraient aussi permettre d'identifier les gènes responsables de tel ou tel effet pathogène.
D'autre part, certaines données peuvent désormais être utilisées en diagnostic ou dans le pronostic du risque de développement d'infections: l'identification de la séquence répétée Ng-rep peut par exemple être utilisée pour détecter une contamination par Neisseria. L'amplification de séquences répétées permet aussi d'identifier une contamination par diverses bactéries, telles que les Chlamydiae, M. tuberculosis, H. influenzae, M. pneumoniae, H. pylori, N. meningitidis...
Plusieurs protéines de bactéries extrêmophiles sont commercialisées, la plus remarquable est la polymérase Taq, dérivée de Thermus aquaticus (thermophile), enzyme clé de la PCR, et très utilisée dans les laboratoires de biologie moléculaire. Une cellulase isolée à partir d'une bactérie alcalophile représente un autre exemple de protéine industriellement utilisée (préparation de coton).
3. Génomes des modèles eucaryotes
3.1 Structure des chromosomes
La levure a été le premier Eucaryote chez lequel tous les chromosomes aient été séquencés. On espérait alors pouvoir détecter quelques formes de régularité à longue distance au sein des séquences de ces chromosomes entiers. Quelques grands traits d'organisation ont effectivement été décelés:
- certains chromosomes apparaissent comme une succession de régions d'environ 150 kb alternativement riches ou pauvres en [G+C]. Cette périodicité est corrélée avec la densité en gènes, plus nombreux dans les régions riches en [G+C]. Mais ces caractéristiques n'ont pas été observées chez tous les chromosomes.
- alors que les brins complémentaires des chromosomes codent généralement un nombre similaire de gènes, le nombre de gènes codés par chacun des brins du chromosome II ou de la région centrale du chromosome VI est significativement différent. Aucune explication satisfaisante n'a pu être proposée à cette observation.
- chez les chromosomes de petite taille (chromosomes I ou VI), la séquence montre que leurs extrémités sont occupées par des éléments subtélomériques essentiellement non codants. Leur fonction éventuelle serait "d'allonger" ces chromosomes afin d'en stabiliser la structure et d'assurer une ségrégation correcte lors des divisions cellulaires.
Chez C. elegans, le génome est remarquablement uniforme du point de vue de la teneur en [G+C], de l'ordre de 36%. Cette teneur ne varie pratiquement pas le long des chromosomes, contrairement à ce qui a été observé chez les mammifères (cf. § 4.4.1). La densité en gènes le long des chromosomes est légèrement plus élevée dans les régions centrales que dans les bras chromosomiques, et plus faible sur le chromosome X.
La taille du génome de la drosophile est de l'ordre de 180 Mb (le chromosome 4 ne contient que 4 Mb). L'hétérochromatine est très large chez cette espèce, elle recouvre environ 60 Mb, et comprend essentiellement des séquences répétées, des éléments transposables, et deux blocs de gènes ribosomiques. Les gènes uniques y sont rares. L'euchromatine couvre 120 Mb, elle contient la majorité des gènes. Elle a été séquencée par Celera selon le principe du séquençage aléatoire: la séquence finale couvre 97% du génome, mais 1.630 lacunes subsistent.
Le génome de la souris est réparti en 20 paires de chromosomes (19 autosomes, et une paire de chromosomes sexuels), tous acrocentriques. La taille globale est environ 14% plus réduite que celle de l'homme, mais la teneur en [G+C] est en revanche très proche (tableau 3), elle est - comme chez l'homme - répartie en régions riches ou pauvres en [G+C].
Le génome d'A. thaliana comprend 5 chromosomes (tous autosomiques) dont la teneur en [G+C] est de l'ordre de 41%. Deux sont acrocentriques (chromosome 4 [18 Mb] et 2 [20 Mb]), deux submétacentriques (chromosome 3 [23 Mb], et 5 [26 Mb]), et un métacentrique (chromosome 1, 29 Mb).
Tableau 3. Caractéristiques des génomes de levure, S. pombe, nématode, drosophile, arabette, et de l'homme. Chez le nématode, la fréquence des gènes est de 4,8 sur les autosomes, 6 sur le chromosome X.
| Levure | Nématode | Drosophile | Arabette | Homme | |
|---|---|---|---|---|---|
| taille physique (Mb) | 13 | 100 | 180 | 125 | 3.000 |
| taille moyenne d'un cM (kb) | 3 | 500 | 300 | 220 | 800 |
| teneur en [G+C] | 38% | 36% | nd | 41% | 41% |
| nombre de gènes | 6.200 | 19.100 | 13.600 | 25.500 | ~30.000 |
| fraction codante | 68% | 27% | 13% | 29% | 1,4% |
| nombre moyen d'exons par gène | 1,04 | 5,5 | 4,6 | 5,2 | 8,7 |
| taille des gènes (kb) | 1,4 | 2,7 | 3 | 2,1 | 28 |
| taille moyenne du codant (introns exclus) | 1.450 | 1.311 | 1.497 | 1.300 | 1.340 |
| taille moyenne des exons (pb) | 1.450 | 218 | 150 | 250 | 145 |
| taille moyenne des introns (pb) | 500 | 267 | 487 | 168 | ~3.300 |
| fréquence des gènes (par kb) | 2 | 4,8 / 6 | 9 | 4,5 | ~100 |
| nombre d'ARNt | 273 | 584 | 284 | 589 | 535 |
| Localisation chromosomique des NOR | 12 | 1 | X et Y | 2, 4 | 13, 14, 15, 21, 22 |
3.2 Identification des gènes
Le première observation remarquable qui découle de l'analyse des séquences de ces organismes est la densité élevée en gènes (tableau 3), supérieure à ce que l'ensemble des observations antérieures laissait supposer.
Chez S. cerevisiae, environ 6.200 gènes ont été identifiés (sans compter les ARNt et les gènes de moins de 300 bases), soit cinq fois plus que ce à quoi on s'attendait. Ceci est corrélé au fait que beaucoup de ces gènes ne se manifestent pas par un phénotype directement observable: ils ne sont par conséquent pas décelables par les mutations les affectant, et n'avaient donc pas été identifiés pas les techniques génétiques conventionnelles. Des gènes recouvrants ont été mis à jour, tels que SMD1 et PRP38, localisés sur les brins opposés de l'ADN chromosomiques, et dont les régions 3' codantes se recouvrent.
Le génome de S. pombe comporte environ 4.900 gènes. Alors que peu de gènes sont fragmentés en plusieurs exons chez la levure (moins de 5%), cette fragmentation est plus fréquente chez S. pombe, puisqu'elle concerne 43% des gènes (le nombre d'exons le plus élevé observé chez un même gène est de 16).
Chez C. elegans, la densité en gènes prédits sur le génome a aussi dépassé tous les pronostics: 19.100 gènes ont été identifiés. Chez cette espèce, le séquençage génomique a révélé un nombre étonnamment élevé de gènes localisés dans l'intron d'un autre gène, ainsi que de gènes recouvrants. Plus inattendu encore a été la mise en évidence d'une fréquente organisation de ces gènes en opérons: plus de 15% des gènes du nématode s'organisent ainsi, alors que l'on pensait que ce type d'agencement ne se rencontrait que chez les Procaryotes.
Le nombre de gènes identifiés chez D. melanogaster est de 13.600. Dans ce cas cependant, il était surprenant que ce chiffre soit inférieur à celui obtenu chez le nématode: la drosophile est en effet un Métazoaire triblastique coelomate, traditionnellement considéré comme représentant un stade d'évolution plus avancé que celui du nématode, qui est un pseudocoelomate. Et une drosophile comprend 10 fois de cellules que le nématode, et présente des comportements bien plus évolués (elle vole !).
Chez la souris, le nombre total de gène est de l'ordre de 27.000 - 30.500, fourchette tout à fait comparable à l'estimation obtenue chez l'homme. Plus de 98% d'entre eux sont d'ailleurs homologues à un gène humain. Aucun opéron n'a jamais été observé, ni chez la souris, ni chez aucun autre Vertébré (à ce jour, des opérons eucaryotes n'ont été observés que chez quelques espèces de Némathelmintes - dont fait partie C. elegans - ou de Plathelminthes - tel que par exemple Schistosoma mansoni).
Chez A. thaliana, 25.500 gènes ont été identifiés. Ce chiffre est supérieur à ceux obtenus chez le nématode ou la drosophile. Mais une telle comparaison doit être prudente: d'une part les épissages alternatifs sont peu fréquents chez A. thaliana (moins de 5% des gènes, contre 20-35% chez les Métazoaires). Ce génome a d'autre part récemment subit une tétraploïdisation (duplication globale du génome, fréquente chez les végétaux), ce qui rend probablement compte de ce nombre élevé de gènes (ce qui se retrouve dans le nombre de gènes codant des ARNt, plus élevé que chez toute autre espèce eucaryote entièrement séquencée).
3.3 Fonctions des gènes reconnus ou prédits
Pour un certain nombre de gènes chez toutes ces espèces, l'analyse des séquences protéiques déduites des séquences nucléotidiques permit - par comparaison avec les gènes déjà connus - de prédire leur fonction. Grâce au séquençage systématique, le nombre de gènes potentiellement impliqués dans une fonction biologique donnée s'est donc soudainement accru, et ceci beaucoup plus rapidement que si des moyens de recherche classique avaient été utilisés. Il subsiste cependant une fraction importante de gènes pour lesquels aucune parenté avec des gènes déjà fonctionnellement connus n'apparaît (40 à 60%, selon les espèces).
Le séquençage d'un génome peut révéler des fonctions insoupçonnées chez un organisme: chez la levure par exemple, a été identifié un gène codant une histone H1, dont l'existence n'était pas supposée auparavant. Parmi les gènes trouvés uniquement chez le nématode, certaines codent des protéines SXC, impliquées dans des interactions avec la matrice extracellulaire. Chez A. thaliana a été identifié le gène de lyase hydroxynitrile, produisant de l'HCN, répulsif pour les herbivores.
Beaucoup des gènes impliqués dans la division cellulaire ne sont pas conservés chez tous les Eucaryotes séquencés: les gènes codant les cyclines de la levure n'ont par exemple pas d'équivalent chez les multicellulaires. A l'inverse, les cyclines de la drosophile, du nématode, ou des Vertébrés, sont apparentées.
Chez la drosophile ou le nématode, un nombre important de gènes codent des protéines impliquées dans le cytosquelette (actine, tubuline...) ou dans la motricité (myosine, dynéine...). Ces gènes représentent des homologues de familles présentes chez les Vertébrés.
L'importance de l'ADN transcrit mais non traduit a été établie chez toutes ces espèces. Elle comprend en particulier les gènes codant les ARNt, les ARNr, qui forment les organisateurs nucléolaires, localisés dans différentes régions chromosomiques, et les 5S. Curieusement, 40% des gènes codant des ARNt sont situés sur le chromosome X chez le nématode.
3.4 Régions non codantes
La répartition des séquences répétées chez les organismes modèles - ainsi que chez l'homme - est représentée dans le tableau 4. Sur l'ensemble du génome, elle atteint près de 15% chez la drosophile (en raison de l'abondance de l'hétérochromatine); mais cette fraction est toujours nettement plus faible que chez l'homme.
Tableau 4. Fréquences des séquences répétées (entre parenthèse: fréquence chez l'euchromatine et l'hétérochromatine chez la drosophile)
| Arabette | Nématode | Drosophile | Souris | Homme | |
|---|---|---|---|---|---|
| LINE/SINE | 0,5% | 0,4% | 4,7% (0,7% + 13,2%) | 28% | 28% |
| Séquences type rétrovirus | 4,8% | 0% | 6,4% (1,5% + 16,9%) | 10% | 7 % |
| Séquences type transposons | 5,1% | 5,3% | 3,6% (0,7% + 9,9%) | 1% | 3% |
| Total | 10,5% | 6,5% | 14,9% (3,1% + 40,2%) | 38% | 38% |
Chez C. elegans, diverses familles de séquences répétées, en tandem ou dispersées, ont été mises en évidence. Certaines de ces familles sont préférentiellement situées dans des introns, d'autres en sont exclues. D'autres biais de localisation sont observés par rapport aux divers autosomes et chromosome X, ou entre bras autosomiques et régions centrales.
Le génome d'A. thaliana contient un faible nombre de séquences répétées, de l'ordre de 10% (tableau 4), et cette fraction est très faible par rapport à celle connue chez les autres plantes. Dans les régions centromériques ou péri-centromériques, le nombre d'éléments transposables et de séquences répétées est élevé, avec une répartition distincte des différentes familles de ces séquences. À l'inverse, le nombre de gènes est faible, et bien que beaucoup d'entre eux ne soient pas fonctionnels, une fraction est toutefois transcrite, et certains correspondent à des gènes de fonction connue (par exemple des ATPases, des transporteurs ABC, des enzymes de réplication de l'ADN, des hélicases...)
La souris représente la deuxième espèce de mammifère (après l'homme, voir ci-dessous) dont le séquençage global ait été entrepris. Dans ces deux cas, le travail de séquençage est handicapé par l'abondance des séquences répétées, car - de par leur ressemblance - différentes séquences répétées obtenues à partir d'un clone génomique peuvent être assemblées alors qu'elles proviennent de régions distinctes.
Deux types de séquences répétées sont distinguées : les séquences répétées en tandem et séquences répétées dispersées. Les séquences répétées en tandem comprennent les microsatellites, constitués de répétitions de motifs dont la taille peut varier entre 1 et 13 nucléotides (fig. 2). Ils sont fréquemment polymorphes, et distribués uniformément le long du génome. Ces séquences ont été en particulier utilisées pour établir les cartes génétiques (voir chapitre 2). Un autre ensemble de séquences répétées en tandem est représenté par les minisatellites, dont la longueur du motif de base est de 14 à 500 paires de bases. Leur répétition peut s'étendre sur 0,5 - 30 kb.
Figure 2. Exemples de séquences dADN satellite (souligné). A: microsatellite: la taille du motif répété peut varier entre lunité et la dizaine de nucléotides. Le microsatellite représenté ici est une répétition de 25 dinucléotides CA. B: minisatellite: la taille du motif répété peut aller de la dizaine à la centaine de nucléotides. Le motif est ici une séquence CGGGCAGGAGGGGGAGG (dont la répétition nest pas strictement identique dun motif à lautre).
A
.......G C T G A G C C G G C T C C T G A G A G A A G C G C T T T C T G A G T C G T T T C G A G G A C A G C C C T G G C C G G T C T T T C C A G G C T G T G A G G G G C T C C T G G G A C T G C T G T C T C C T C T T A T C C T G T A C C T C T G C C A T G T G A C A C A C A C A C A C A C A C A C A C A C A C A C A C A C A C A C A C A C A C A C A C A C A C A C A T A A A T T A T C C T G G A G G A A A G G T T A A G G T G A C A C A T G G A G A C T G A G T G T C A C C G T T A T T T C C G C A G G T C C T C T C T G A T G A C A T G A A G A A G C T G A A G G C C C G A A T G G T A A T G C T C C T C C C T A C T T C T G C T C A G G G G T T G G G G G C C T G G G T C T C A G C G T G T G A C A C T G A G G A C A C T G T G G G A C A C C T G G G A C C C T G G A G G G A C A A G G A T C C G G CC C T T
B
.......T C A G G G T G A G A A G G A T G A A A A G G G A C C C A C A G G C T C C C T C A C C C C T T A C C G T G G G C A A A T G C T T G C A C C T G G G T G G C A G T G A G T G G G C G G G T A A T C G G G C A G G A G G G G G A G G C G G G C A G G A G G G G G G A G G C G G G C A C G A G G G G G G A G G T G A G C A G G A G G G G G A G G C G G G C A G G A G G A G G A G G C G G G C A G G A G G G G G A G A C G G G C A G G A G A G G G A G G C A G G C A G G A G A G G G A G G T G G G C A G G A G G G G G G G G C G G G C A G G A G G A G G A G G T G G G C A G G A G G G G G A G G C G G G C A G G A G G G G G A G G C G G G C A G G A G G G T G A G G G G G G A T C T G G A C G C C C G G G G A G A C T G A G G G A G G C A T C C A A G C C C C A G G G C T C C T T G A G G A A A C A A C A G G G G T G C C A G A C G T G G C C C G G G C C C C T G G C T G G G C C C A G T T C G G G G T G T G T G G G A G C T G A G G A C T C A C T G G G C T T G A G G A C T G A C T G A T G T G G A.....
Les séquences répétées dispersées sont représentées par les SINE, les LINE, les rétroposons à LTR, et les transposons à ADN. Ils dérivent d'éléments transposables, les trois premiers transposant via l'ARN, le quatrième via l'ADN.
Les LINE et les SINE présentent une distribution complémentaire dans le génome: les SINE prédominent dans les isochores riches en [G+C], alors que les LINE prédominent dans les isochores pauvres en [G+C]. Globalement, les séquences répétées dispersées couvrent près de 40% du génome murin.
4. Génome humain
4.1 Les chromosomes humains
La longueur totale du génome humain est d'environ 3.000 Mb. Le séquençage global de ce génome (qui n'est pas terminé) a impliqué une collaboration internationale entre plus de 20 laboratoires de 6 pays (USA, Grande-Bretagne, Japon, France, Allemagne et Chine). Les séquences étaient (et sont toujours) d'accès libre et immédiat, assemblées et orientées grâce aux données de cartographie génétique et physique, et de comparaisons de séquences. La montée en puissance de ce programme international est une conséquence des importants progrès technologiques et informatiques, qui ont par exemple permis d'atteindre une vitesse globale de séquençage de 1.000 nucléotides par seconde. Aujourd'hui 96% du génome humain est totalement séquencé et plusieurs chromosomes sont terminés (tableau 5).
Tableau 5. Chromosomes humains complètement séquencés.
| Chromosome | caryotype | taille | fraction du génome | contenu en [G+C]; | lacunes | nombre de gènes |
|---|---|---|---|---|---|---|
| 6 | sumétacentrique | 166,9 Mb | 5,6 % | 40 % | 6 | 2190 (dont 633 pseudogènes) |
| 7 | sumétacentrique | 154 Mb | 4,8 % | 41 % | 10 | 2091 (dont 941 pseudogènes) |
| 9 | sumétacentrique | 109 Mb | 3,6 % | 41,4 % | 5 | 1575 (dont 426 pseudogènes) |
| 10 | sumétacentrique | 131,7 Mb | 4,4 % | 41,6 % | 9 | 1787 (dont 430 pseudogenes) |
| 13 | acrocentrique | 95,6 Mb | 3,2 % | 38,5 % | 6 | 929 (dont 296 pseudogènes) |
| 14 | acrocentrique | 87,4 Mb | 2,7% | 40,9 % | 0 | 1128 (dont 292 pseudogènes) |
| 19 | métacentrique | 56 Mb | 1,9 % | 48 % | 3 | 1782 (dont 321 pseudogènes) |
| 20 | métacentrique | 59,1 Mb | 1,8% | 44,1 % | 4 (~320 kb) | 895 (dont 168 pseudogènes) |
| 21 | acrocentrique | 33,5 Mb | 1,0 % | 40,9 % | 3 (~100 kb) | 284 (dont 59 pseudogènes) |
| 22 | acrocentrique | 33,5 Mb | 1,0 % | 47,8 % | 11 (~150 kb) | 679 (dont 134 pseudogènes) |
| Y | acrocentrique | 60 Mb | 1,8% | nd | 4 | 156 (nombreux pseudogènes) |
Plusieurs observations antérieures ont été réexaminées à l'aide de ces nouvelles données. Bernardi avait par exemple montré (par fractionnement d'ADN génomique sur gradient de sulfate de césium) que la distribution en [G+C] chez l'homme est loin d'être uniforme: le génome est une mosaïque de régions de diverses compositions en [G+C], appelés isochores. Cinq isochores ont été reconnues: les isochores L (Light) pauvres en [G+C]: L1 ([G+C] < 38%) et L2 (38% < [G+C] < 42%), et les isochores H (Heavy) riches en [G+C]: H1 (42% < [G+C] < 47%), H2 (47% < [G+C] < 52%), et H3 ([G+C] > 52%). Les isochores H correspondent aux bandes R obtenues par coloration au Giemsa, les isochores L aux bandes G. Cette hétérogénéité est observée sur tous les génomes d'homéothermes (oiseaux et mammifères), mais chez ceux des poïkilothermes.
4.2 Identification des gènes
Le génome humain porte 535 gènes codant des ARNt, nombre plus faible que chez le nématode, mais plus élevé que chez la drosophile (tableau 4). Près d'un quart de ces gènes sont localisés sur les chromosomes 1 et 6 (le chromosome 22 n'en contient aucun). Les gènes codant les ARNr 18S, 28S, et 5,8S sont organisés en 150-200 groupes de 44 kb, répartis sur les chromosomes 13, 14, 15, 21 et 22. Enfin, le génome contient environ 2.000 gènes ribosomiques 5S, dont une fraction importante est localisée sur le chromosome 1.
Les gènes codants des protéines ont été prédits par (i) comparaison aux bases de données d'EST, (ii) comparaison aux séquences complètes d'ARNm, (iii) et des programmes de prédiction tel que GENSCAN. Le nombre total de gènes chez l'homme demeure à ce jour incertain, il oscille entre 26.000 et 35.000. Ces chiffres étaient inattendus: l'homme n'aurait donc que deux fois plus de gènes que la drosophile ou le nématode. La répartition globale des gènes est de 11,1 gènes / Mb, le chromosome le plus riche en gènes est le 19 (26,8 gènes/Mb), les plus pauvres sont le 13 et le Y (6,4 gènes/Mb). Certaines régions - de plus de 500 kb - ne contiennent aucun gène, elles recouvrent 605 Mb, soit 20% du génome. La plus grande recouvre 3 Mb sur le chromosome 13.
Les isochores ne présentent pas une densité en gènes identique: les isochores H sont plus riches en gènes que les isochores L. Les chromosomes 17, 19 et 22, qui comportent beaucoup d'isochore H3, ont par exemple une densité en gènes élevée, les chromosomes X, 4, 18, Y, qui en comportent peu, ont une densité en gènes faible. Mais cette répartition n'est pas uniforme: le chromosome 15, qui a un nombre d'isochore H3 faible, a une densité en gènes normale, et le chromosome 8, qui a un nombre d'isochore H3 normal, a une densité en gènes faible.
La taille moyenne des gènes transcrits est de 27.900 pb. La fraction codante couvre généralement 1.340 pb, répartis en 8-9 exons, et la taille moyenne des exons est de 145 pb (généralement entre 50 - 200 pb, seulement 42 exons ont une taille inférieure à 19 pb). La taille moyenne des introns est d'environ 3.300 pb. Plus de 35% des gènes présentent un épissage alternatif. Globalement, 28% du génome serait transcrit en ARNr, ARNm, ARNt, ou ARN de petites taille non traduits, seulement 1,4 % serait traduit.
Le gène de plus grande taille est celui codant la dystrophine, qui s'étend sur 2,4 Mb. Le gène de la titine présente d'autres records: il code le plus grand messager humain connu (80.780 pb), le plus important nombre d'exons (178), parmi lesquels figure le plus grand exon connu (17.106 pb).
Une première évaluation de la diversité protéique peut être obtenue par l'analyse des domaines protéiques: environ 51% des protéines comportent au moins un domaine identifiable, et au total 1.262 familles de domaines ont été répertoriées chez l'homme. Cette diversité des domaines est plus élevée que chez les autres Eucaryotes à ce jour séquencés: 1.035 familles ont été identifiés chez la drosophile, 1.014 chez le nématode, 851 chez la levure, et 1.010 chez Arabidopsis.
Toutefois, un faible nombre de domaines protéiques est spécifique des Vertébrés: seulement 94 d'entre eux (soit 7%) ne sont détectés chez aucune autre espèce non-Vertébré: peu de domaines ont donc été "inventés" dans ce groupe, la majorité est présente chez d'autres Métazoaires. Un seul de ces domaines Vertébré-spécifiques est présent chez un gène codant un enzyme, ce qui conforte l'idée que l'origine des protéines enzymatiques est ancienne, et que la majorité d'entre elles sont partagées par l'ensemble du vivant.
Mais si l'invention de domaines est faible chez les Vertébrés, la diversité des architectures protéiques humaine est élevée. Une architecture se défini comme l'organisation linéaire de différents domaines le long d'un polypeptide. L'homme contient 1,8 fois plus d'architectures différentes que la drosophile ou le nématode, 2,9 fois plus que A. thaliana, 5,8 fois plus que la levure. Une autre diversification protéique est due à l'expansion de certains domaines: 60% des domaines sont présents chez plus de protéines chez l'homme que chez tout autre Eucaryote. Citons le domaine FGF (observé dans 30 protéines chez l'homme, seulement 2 chez la drosophile ou le nématode) ou TGF-ß (42 chez l'homme, 9 chez la drosophile, 6 chez le nématode).
Certains domaines présentent une expansion remarquable chez l'homme : le domaine de type immunoglobuline (absent chez Arabidopsis et la levure) est par exemple très diversifié. Il n'est présent chez les Invertébrés que chez quelques protéines de surface, alors que chez les Vertébrés il est présent chez de nombreuses protéines du système immunitaire, telles que les immunoglobulines, les récepteurs des lymphocytes T, des protéines de surface lymphocytaires (telles que par exemple les protéines d'histocompatibilité)... Ce qui illustre la diversité qu'un domaine permet d'obtenir, concernant dans ce cas le système immunitaire. Des observations similaires sont observées pour des domaines associés à des protéines du système nerveux, avec par exemple les domaines observés chez les neurotrophines et leurs récepteurs, ou les protéines de signalisation. Cette diversification est dans ce cas liée à celle de notre système nerveux : diversité des types de cellules neuronales ou gliales, des jonctions synaptiques, des types de transmission...
La diversité architecturale se manifeste aussi par la diversité des associations entre domaines, significativement plus marquée chez l'homme que chez la drosophile, le nématode ou Arabidopsis. Le domaine correspondant à la protéase de sérine de type trypsine est par exemple associée à 18 autres domaines différents dans diverses protéines chez l'homme, mais seulement 8 autres chez la drosophile, 5 chez le nématode, un seul chez la levure.
Pour environ 60% des protéines, leur fonction est connue ou peut être suggérée. Les plus abondantes concernent les protéines impliquées dans la régulation de la transcription, le métabolisme nucléique, les récepteurs de signaux. Mais la fonction protéique demeure inconnue pour plus de 40% d'entre elles.
Nous avons vu que le nombre de gènes humains est environ deux fois supérieur à celui de la drosophile ou du nématode. Ceci peut être illustré par exemple par les gènes homéotiques, représentés par environ 220 exemplaires chez l'homme, mais seulement 90 chez la drosophile ou le nématode (une dizaine chez la levure).
La diversité protéomique de l'homme se manifeste donc par le nombre de gènes présents, le nombre de domaines protéiques et la multiplicité de leurs architectures. Elle se manifeste aussi par les épissages alternatifs, la diversité des systèmes de régulation de la transcription, des interactions inter-protéiques, des modifications post-traductionnelles, caractéristiques entrant dans le cadre du transcriptome ou du protéome (cf. chapitres 5 et 6).
4.3 Séquences répétées
Le génome humain est le premier contenant une proportion élevée de séquences répétées dont le séquençage a été entrepris, celle-ci est bien plus élevée que dans le génome d'Arabidopsis, du nématode, ou de la drosophile.
L'organisation des LINE, SINE, rétroposons à LTR, et transposons à ADN a été décrite la dernière partie du § 4.3.7, concernant la souris, et d'autres informations sont données dans le tableau 3. Trois familles de LINE existent chez l'homme: LINE1, LINE2, et LINE3 (seul le premier est encore actif). Une quarantaine de familles de SINE a été identifiée. Les représentants les plus fréquents sont les séquences Alu, les MIR (Mammalian wide-Interspersed Repeat), et les MER (MEdium Reiteration frequency repeats). Les rétroposons à LTR et les transposons à ADN sont aujourd'hui tous inactifs.
Comme chez la souris, les séquences LINE et SINE présentent une distribution complémentaire dans les isochores riches ou pauvres en [G+C]. Les rétroposons à LTR et les transposons à ADN sont répartis de façon uniforme. L'ensemble des séquences répétées dispersées couvrent environ 40% du génome humain (tableau 3).
Certaines régions sont très riches en séquences répétées dispersées: une région de 525 kb du chromosome Xp11 comprend par exemple 89% de répétées dispersées. A l'inverse, des régions très pauvres en séquences répétées dispersées sont par exemple représentées par celles contenant les gènes Hox, ou par des régions très pauvres en gènes: une région de 100 kb du chromosome 1 ne contenant aucun gène, ne contient que 5% de répétées.
Conclusion
Ces dernières années, le séquençage des génomes de Procaryotes s'est révélé particulièrement fécond. Un génome bactérien est séquencé tous les deux mois, et à ce jour, une centaine de séquences complètes de génomes bactériens ont été obtenues. L'importante diversité de ces génomes, et des gènes qu'ils contiennent, a ainsi été mise en évidence: pour la moitié d'entre eux, la fonction de la protéine codée reste inconnue.
L'identification de la fonction de gènes chez A. thaliana a déjà eut des retombées chez les espèces cultivées. Ont par exemple été identifiés chez A. thaliana des gènes impliqués dans la libération des graines, ce qui a permis d'améliorer la production chez le colza, ou des gènes responsables de la maturation des fruits, ce qui a permis d'augmenter la production de la tomate. Par ailleurs, deux enzymes impliqués dans la production de lipides poly-insaturés ont été identifiées chez A. thaliana. Les homologues ont été identifiés chez le soja, et chez une variété transgénique de cette espèce, chez laquelle l'expression de l'un de ces deux gènes a été supprimée, l'abondance des lipides mono-insaturés est passée de 25% à 85%, et celle des poly-insaturés sont passées de 60% à 2%. Cet exemple d'application des recherches menées chez A. thaliana reflète les retombées que l'on peut attendre des travaux réalisées chez cette espèce.
Le séquençage du génome humain représente un programme sans précédent, qui représente 25 fois celui réalisé chez la drosophile, 8 fois la totalité des génomes séquencés. Il devrait permettre des progrès médicaux importants, puisqu'il permet une approche systématique et non biaisée du génome. L'identification de gènes responsables de maladies génétiques monogéniques ou plurifactorielles en est par exemple largement facilité. Globalement, la connaissance de ce génome, les études des gènes présents, devraient permettre d'accéder à une meilleure compréhension de leur fonction, des stades de développement, du métabolisme, des résistances aux pathogènes.
"Analyse de Génomes, Transcriptomes et Protéome"
Alain Bernot
Ed. Dunod
 home |
présentation |
ressources |
séquençage |
actualités |
presse |
site |
mentions légales
home |
présentation |
ressources |
séquençage |
actualités |
presse |
site |
mentions légaleswebmaster@genoscope.cns.fr